Chemistry
The search for a small molecular inhibitor of the HCV NS3 protease began by screening compound libraries. Traditional high-throughput screening of compound libraries failed to identify chemical leads that could be followed-up by optimization of medicinal chemistry structure activity. Given no viable lead from screening, a structure-based design approach was adopted. The first compounds synthesized were based on an HCV protease substrate, incorporating reversible electrophilic traps that could potentially bind covalently to catalytic Ser139. Introducing a ketoamide moiety generated an undecapeptide (Analog 1) (Fig. 4) that inhibited NS3 enzyme activity at low nanomolar concentrations (Ki* = 1.9 nmol/L).
However, this lead compound lacked almost all the desired drug-like characteristics, and was unsuitable for clinical development, as it contained multiple peptidic bonds derived from natural amino acids, had a high molecular weight, and was metabolically unstable. Although no specific target molecular weight was defined in these studies, general experience suggests that lower molecular weight compounds have improved permeability and tend to be more easily absorbed. Optimization of this undecapeptide initially focused on reducing its molecular weight. The initial SAR was derived primarily through directed combinatorial synthesis and information derived from the NS3 X-ray structure. Highly modified non-proteinogenic amino acids were also introduced in an attempt to improve ligand efficiency and metabolic stability. After considerable optimization, a leucine P2-derived pentapeptide (Analog 5) spanning P3–P2′ was identified, which retained significant activity against the NS3 enzyme (Ki* = 630 nmol/L) (Fig. 4). This was one of the first analogs to demonstrate potency in the sub-micromolar range. Replacement of leucine with cyclopropylalanine at P2 resulted in Analog 6,which exhibited improved enzyme-binding potency(Ki* = 50 nmol/L, Fig. 4).29
Most of the initial SAR studies during the early stages were guided by enzyme-binding assays because of the lack of robust cellular assays. The introduction of the replicon cell-based assay was pivotal in further refining SAR, and allowed identification of compounds that were active in both binding and cellular assays. Unfortunately, Analog 6 had a half-maximal effective concentration (EC50) of >5000 nmol/L in the replicon-based cellular assay, despite its promising enzyme-binding affinity. It was unclear how to interpret these results. One hypothesis was that Analog 6 was polar, containing many amide bonds that prevented it crossing cell membranes. By introducing peptide isosteres, modifying the side chains of amino acids, and methylating amide nitrogens to depeptidize the molecule, the methylated Analog 7 was identified; this demonstrated similar solubility and enzyme activity to Analog 6 (Ki* = 60 nmol/L) but with much improved cellular activity in the replicon assay (EC90 = 950 nmol/L). Comparison of Analogs 6 and 7 revealed that addition of a secondary amide at P2 yielded compounds that retained good enzyme activity and exhibited favorable potency in the replicon-based cell-culture assay. Further optimization of this secondary amide was investigated via the addition of proline analogs at P2 (Fig. 4).
Cyclization of the N-methyl group to a P2 cyclopropyl group generated the cyclopropylated Analog 8, which had greater enzyme-binding affinity (Ki* = 11 nmol/L) than Analog 6. Introduction of a gem dimethyl group at the cyclopropyl proline resulted in Analog 9, and was associated with improved enzyme-binding affinity and increased potency in the replicon-based cell-culture assay (Ki* = 1.4 nmol/L; EC90 = 90 nmol/L).30
Having established that Analog 9 showed desirable binding and cellular activities, its oral pharmacokinetics were next evaluated. In rats, administration of Analog 9 (10 mg/kg) was associated with poor plasma exposure, with an area under the plasma concentration curve (AUC) of 0.23 μmol/L/h and oral bioavailability of 1%. These data indicated that the compound was poorly absorbed, suggesting that it was peptidic and needed further reduction in molecular weight. To reduce molecular weight to an acceptable level and improve oral pharmacokinetics, a series of compounds were synthesized that were truncated at P1′ with small groups. Capping compounds similar to Analog 9 at P1′ with a primary amide improved oral exposure (Fig. 4).
Truncation of the Analog-9 containing norvaline at P1′ with a primary amide (to produce Analog 11) significantly reduced enzyme activity (Ki* = 100 nmol/L). However, this analog had improved oral absorption resulting in increased oral exposure (AUC = 2.52 μmol/L/h). Thus, truncating compounds at P1′ as a primary ketoamide improved bioavailability but detrimentally affected binding affinity. Modifications at P1, at P3, and at P3 caps were therefore reconsidered to identify groups that make stronger lipophilic contact with the enzyme, consistent with an improved binding profile. A systematic modification of P1 by the introduction of carbocyclic alanine derivatives led to the discovery of a cyclobutyl methyl group as the optimal P1 moiety based on its excellent contact at the S1 pocket. The resulting Analog 12 exhibited favorable enzyme-binding and cellular activity (Ki* = 8 nmol/L; EC90 = 700 nmol/L). In addition, Analog 12 also demonstrated improved oral pharmacokinetics with an AUC of 1.5 μmol/L/h when administered at a dose of 10 mg/kg. Finally, P3 SAR modifications in combination with changes at the P3 capping group identified tert-butyl glycine at P3 and tert-butyl urea at the P3 cap, resulting in Analog 13 (SCH503034, boceprevir)31; this had improved selectivity against human neutrophil elastase, and retained activity in enzyme and cellular replicon-based assays. The molecular interactions between boceprevir and the NS3/4A protease are depicted in Fig. 5 (A,B).
In vitro characterization of boceprevir
Evaluation of boceprevir in the continuous enzyme assay revealed time-dependent inhibition of genotypes 1–6 NS3/4A proteases in a progress curve analysis yielding overall Ki values of 10–104 nmol/L (Table 1).32–34 Since mammalian serine proteases perform many important biological functions, especially in relation to digestion, blood clotting, and complement cascade during antibody neutralization, boceprevir was evaluated against a panel of other human proteases (e.g. human neutrophil elastase, human neutrophil cathepsin G, and human liver cathepsins H and L). Boceprevir was found to be selective for inhibition of the HCV protease. A selectivity ratio for the HCV NS3 protease ranging from 4 to >7000 was observed when boceprevir was evaluated against a number of human serine and cysteine proteases (Table 1).
Table 1In vitro characteristics and preclinical pharmacokinetics of boceprevir
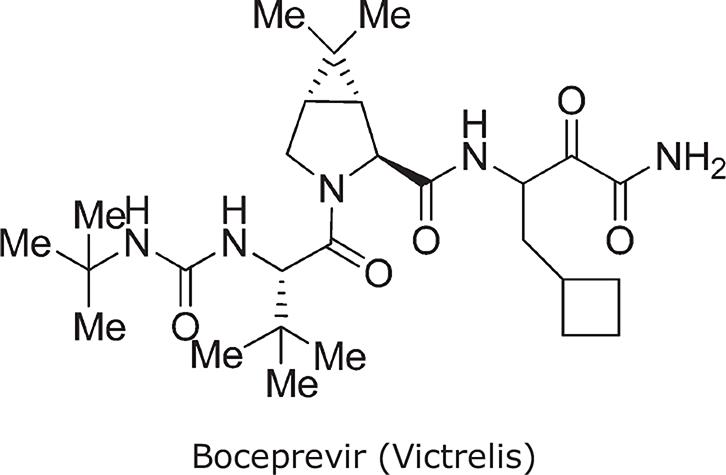 |
| Chemical name | ((1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide) |
| Molecular weight | 519.7 |
| HCV | NS3/4A enzyme,34Ki* ± SD (nmol/L) | Replicon34EC50± SD (nmol/L) |
| GT 1a | 23 ± 9 (n = 18) | 236 + 56 (n = 21) |
| GT 1b | 22 ± 5 (n = 20) | 219 + 47 (n= 16) |
| GT 2a | 26 ± 13 (n = 6) | 317 ± 31 (n = 3) |
| GT 2b | 6 ± 1 (n = 10) | 180 ± 10 (n = 3) |
| GT 3a | 17 ± 4 (n = 14) | 353 ± 133 (n = 3) |
| GT 4a | 104 ± 18 (n = 18) | n/a |
| GT 5a | 53 ± 15 (n = 12) | 250 ± 78 (n = 3) |
| GT 6a | 10 ± 2 ( n = 6) | n/a |
| Therapeutic Indexa | ≥20 |
| Selectivity Indexb | 4 to ≥7000 |
| Bioavailability (rat) | 26% |
| Bioavailability (dog) | 30% |
| Bioavailability (mouse) | 34% |
| Bioavailability (monkey) | 4–11% |
| Liver/plasma ratio (rat) | Approximately 30 at 10 mg/kg |
| CYP 2D6, 2C9, 2C19 | >30 μmol/L |
| CYP 3A4 | >30 μmol/L | 8.5 μmol/L |
In the HCV replicon cell assay, boceprevir potently reduced HCV RNA levels with EC50 values of 200–400 nmol/L in genotypes 1, 2, and 5 replicons (Table 1).32 Incubation of replicon-bearing Huh7 cells for 72 hours with 2.5 μmol/L boceprevir reduced HCV RNA level by 1.5 logs, and prolonged exposure for up to 14 days resulted in replicon RNA levels falling by approximately 4.0 logs, equivalent to <1 copy of HCV RNA remaining per cell.32 No cytotoxicity was observed in human hepatoma cells or stimulated peripheral blood mononuclear cells.